XANESNET

XANESNET is a machine learning code to support the analysis of X-ray spectroscopy. It is freely available and can be downloaded from here. An original version of the code implemented using Keras rather than PyTorch can be found here.
The code has the following capabilities:
XANESNET is under continuous development, so feel free to flag up any issues/make pull requests - we appreciate your input!
The code has the following capabilities:
- Models can operate in both the forward (property/structure-to-spectrum) and reverse (spectrum-to-property/structure) directions.
- Available architectures include: MLP, CNN and LSTM
- Available models including feed forward networks, auto encoders and generative adversarial networks.
- Available descriptors include: RDC, wACSF, SOAP, MBTR and LMBTR
- Spectra can be treated in real (energy) and Fourier space.
- SHAPs analysis can be performed to analyse models
- Bootstrapping, Monte-Carlo dropout and enabling can be used to estimate model uncertainty.
- Data augmentation.
XANESNET is under continuous development, so feel free to flag up any issues/make pull requests - we appreciate your input!
X-ray spectroscopy Training Sets
A machine learning model is only as good as the training set used to develop it. All of our training sets are publicly available and can be downloaded from here.
For Structure to Spectrum Models: Our reference datasets comprise site geometries (‘samples’) of first-row transition metal (Ti–Zn) complexes harvested from the transition metal Quantum Machine (tmQM) dataset [link]. The dataset for each first-row transition metal comprised all of the structures from the tmQM dataset containing that element, as extracted from the 2020 release of the Cambridge Structural Database (CSD) and subsequently optimised at the GFN2-xTB level of theory. The tmQM dataset was initially generated by applying seven filters to exclude: (i) all structures except those containing a single transition metal; (ii) all structures not containing a minimum of one C and one H atom (allowing only these other elements: B, Si, N, P, As, O, S, Se, F, Cl, Br, and I); (iii) the structure of all extraneous molecular components beyond that of the transition metal complex (e.g. counter-ions); (iv) all polymeric structures; (v) all structures without three-dimensional coordinates; (vi) all structures with disordered atoms; and (vii) all structures with charges greater than +1 and less than −1. Full details of the construction and composition of the tmQM dataset can be found [here].
For the XAS set, the K-edge XANES spectra (“labels”) for these structures were calculated using multiple scattering theory (MST) as implemented in the FDMNES package. We have developed nine independent reference datasets, one for each first-row transition metal (Ti–Zn) x-ray absorption edge; the number of samples contained in the reference datasets scales from as few as ∼1100 (V) to ∼8660 (Ni).
For the XES training data, the VtC-XES spectra (‘labels’) for all of the structures in our reference datasets were calculated using a quasi-one-electron approach implemented in the ORCA quantum chemistry package. All VtC-XES spectrum calculations used the TPSSh exchange and correlation density functional and the def2-SVP basis set. The light-matter interaction was described using the electric dipole, magnetic dipole, and electric quadrupole transition moments. After calculation, each VtC-XES spectrum was broadened using a Voigt function containing Lorentzian and Gaussian components. The Gaussian component reflects the limited experimental resolution of VtC-XES and had a fixed width of 1.5 eV in all cases. The Lorentzian component reflects the effect of core-hole lifetime broadening and is a sum of the 1s and 2p core-hole lifetime broadening for each element, i.e. it is element-dependent.
For Spectrum to Structure Models: Our reference dataset contains 36,657 spectra-structure pairs. The structures are in the form of the wACSF descriptor and can be read as described in the file. This dataset incorporates 77 of the elements from the periodic table and molecules with a coordination number, defined as the number of atoms within 2.5 Angstrom of the absorbing atom, between 2 and 16. The Fe K-edge XANES spectra ("labels") for these structures were calculated using multiple-scattering theory (MST) within the muffin-tin approximation as implemented in the FEFF package. The calculation used a self-consistent potential and full multiple scattering up to a radius of 6 Angstrom around the absorbing atom. After calculations, the absorption cross-sections were resampled via interpolation into 475 points over an energy range of 7112-7160 eV.
This dataset also includes 22 spectra-structure pairs associated with experimental data to assess the performance of the network when applied to experimental data.
For Structure to Spectrum Models: Our reference datasets comprise site geometries (‘samples’) of first-row transition metal (Ti–Zn) complexes harvested from the transition metal Quantum Machine (tmQM) dataset [link]. The dataset for each first-row transition metal comprised all of the structures from the tmQM dataset containing that element, as extracted from the 2020 release of the Cambridge Structural Database (CSD) and subsequently optimised at the GFN2-xTB level of theory. The tmQM dataset was initially generated by applying seven filters to exclude: (i) all structures except those containing a single transition metal; (ii) all structures not containing a minimum of one C and one H atom (allowing only these other elements: B, Si, N, P, As, O, S, Se, F, Cl, Br, and I); (iii) the structure of all extraneous molecular components beyond that of the transition metal complex (e.g. counter-ions); (iv) all polymeric structures; (v) all structures without three-dimensional coordinates; (vi) all structures with disordered atoms; and (vii) all structures with charges greater than +1 and less than −1. Full details of the construction and composition of the tmQM dataset can be found [here].
For the XAS set, the K-edge XANES spectra (“labels”) for these structures were calculated using multiple scattering theory (MST) as implemented in the FDMNES package. We have developed nine independent reference datasets, one for each first-row transition metal (Ti–Zn) x-ray absorption edge; the number of samples contained in the reference datasets scales from as few as ∼1100 (V) to ∼8660 (Ni).
For the XES training data, the VtC-XES spectra (‘labels’) for all of the structures in our reference datasets were calculated using a quasi-one-electron approach implemented in the ORCA quantum chemistry package. All VtC-XES spectrum calculations used the TPSSh exchange and correlation density functional and the def2-SVP basis set. The light-matter interaction was described using the electric dipole, magnetic dipole, and electric quadrupole transition moments. After calculation, each VtC-XES spectrum was broadened using a Voigt function containing Lorentzian and Gaussian components. The Gaussian component reflects the limited experimental resolution of VtC-XES and had a fixed width of 1.5 eV in all cases. The Lorentzian component reflects the effect of core-hole lifetime broadening and is a sum of the 1s and 2p core-hole lifetime broadening for each element, i.e. it is element-dependent.
For Spectrum to Structure Models: Our reference dataset contains 36,657 spectra-structure pairs. The structures are in the form of the wACSF descriptor and can be read as described in the file. This dataset incorporates 77 of the elements from the periodic table and molecules with a coordination number, defined as the number of atoms within 2.5 Angstrom of the absorbing atom, between 2 and 16. The Fe K-edge XANES spectra ("labels") for these structures were calculated using multiple-scattering theory (MST) within the muffin-tin approximation as implemented in the FEFF package. The calculation used a self-consistent potential and full multiple scattering up to a radius of 6 Angstrom around the absorbing atom. After calculations, the absorption cross-sections were resampled via interpolation into 475 points over an energy range of 7112-7160 eV.
This dataset also includes 22 spectra-structure pairs associated with experimental data to assess the performance of the network when applied to experimental data.
Quantics
The QUANTICS package solves the time-dependent Schroedinger equation to simulate nuclear motion by propagating wavepackets. Various algorithms are possible, depending on the system of interest and the accuracy required. The focus of the package is the Multi-Configurational Time-Dependent Hartree (MCTDH) algorithm. The package grew out of the Heidelberg MCTDH Package.
QUANTICS has many new features compared to the older MCTDH packages. The main changes are the addition of the G-MCTDH algorithm and the direct dynamics DD-vMCG method. It also has an implementation of the real wavepacket propagation algorithm for scattering. The code is now Fortran 90 based with full dynamical allocation of memory. Parallelisation using OpenMP and MPI is made in many parts of the code. File structures have been updated. Version numbers are no longer appended to executable names. Automatic spline fits of data to provide potential functions.
The code has the following capabilities:
The QUANTICS package solves the time-dependent Schroedinger equation to simulate nuclear motion by propagating wavepackets. Various algorithms are possible, depending on the system of interest and the accuracy required. The focus of the package is the Multi-Configurational Time-Dependent Hartree (MCTDH) algorithm. The package grew out of the Heidelberg MCTDH Package.
QUANTICS has many new features compared to the older MCTDH packages. The main changes are the addition of the G-MCTDH algorithm and the direct dynamics DD-vMCG method. It also has an implementation of the real wavepacket propagation algorithm for scattering. The code is now Fortran 90 based with full dynamical allocation of memory. Parallelisation using OpenMP and MPI is made in many parts of the code. File structures have been updated. Version numbers are no longer appended to executable names. Automatic spline fits of data to provide potential functions.
The code has the following capabilities:
- Grid-based Wavepacket Propagation
- standard MCTDH
- multi-layer MCTDH (ML-MCTDH)
- MCTDH including Gaussian Basis Functions (G-MCTDH)
- Numerically exact (standard) using SIL integrator
- Numerically exact using real WP propagation
- Gaussian Wavepacket Propagation using vMCG algorithm
- Direct dynamics using DD-vMCG
- Control calculations using either OCT or LCT.
- Eigenvalues and eigenfunctions
- Energy Relaxation to obtain the ground eigenstate
- Improved Relaxation to obtain eigenstates and eigenenergies of ground and/or low lying excited states.
- Direct diagonalisation of a Hamiltonian matrix using Lanczos
- Filter Diagonalisation to find the eigenvalues of a system from a wavepacket propagation.
- Density Matrix propagation for open or closed systems
- Potential energy function fitting to the MCTDH product form using the natural potential algorithm.
- Fitting a vibronic coupling model Hamiltonian.
CharTED-KMC
CharTED-KMC is a Kinetic Monte-Carlo code used to model charge transport in organic semi-conductors. The source code can be found here
The code has the following capabilities:
The code has the following capabilities:
- Transport of electrons, holes and excitons.
- Interplay between multiple excited states on each site
- Miller-Abrahams and Marcus rates
- Order parameters to control relative orientation and order within thin film.
VCMaker

VCMaker provides a tool that links quantum chemistry calculations to quantum dynamics simulations, with a particular focus on the easy development and assessment of linear vibronic coupling Hamiltonians. The code can be obtained from here.
The code has the following capabilities:
The code has the following capabilities:
- VConverter is a set of utilities that allows for easy conversion of the Hessian and Gradient between outputs generated by quantum chemistry codes and VCMaker format.
- VCMaker generates and assesses linear vibronic coupling Hamiltonians and well as generate displacement scans along normal modes.
XWave
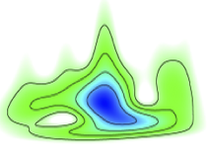
Usually, the first step in the analysis of EXAFS spectra is using a Fourier transform (FT) of the EXAFS signal which yields a pseudo-radial distribution. However, for systems which contain many scattering paths of different atomic species, and/or single and multiple scattering pathways which contribute to the same region of R-space, an unambiguous assignment of the peaks can be difficult. This is because these overlapping scattering pathways will alter the appearance of the FT and give rise to peaks which derive from a combination of multiple pathways, rather than one specific pathway.
XWave is a code, based upon this paper, which performs a Wavelet transform (WT) analysis of EXAFS spectra. Here, the infinitely expanded periodic oscillations of the FT are replaced by a local function, a wavelet, which enables one to analyse the components in k- and R space, concurrently. This yields a 2D correlation plot in both coordinates (analogous to a time-frequency correlation plot) and will separate the contributions between different scattering pathways at the same distance from the absorbing atom and between the contributions of SS and MS events because they will display different k dependencies.
The code is freely available upon request at tom.penfold'at'newcastle.ac.uk.
XWave is a code, based upon this paper, which performs a Wavelet transform (WT) analysis of EXAFS spectra. Here, the infinitely expanded periodic oscillations of the FT are replaced by a local function, a wavelet, which enables one to analyse the components in k- and R space, concurrently. This yields a 2D correlation plot in both coordinates (analogous to a time-frequency correlation plot) and will separate the contributions between different scattering pathways at the same distance from the absorbing atom and between the contributions of SS and MS events because they will display different k dependencies.
The code is freely available upon request at tom.penfold'at'newcastle.ac.uk.